The R class R programming for biologists
Challenges
The sea cucumber challenge
For this challenge, you are going to use what you have learned in the first few weeks to solve common issues when dealing with data, and perform a quick exploration of the dataset. I tried to build the questions so that if you get stuck, you can move on to the following questions.
I am providing you with two datasets:
holothuriidae-specimens.csvcontains a list of sea cucumber specimens housed at natural history museums across the United States. It’s a simplification of a dataset I obtained through the iDigBio portal.holothuriidae-nomina-valid.csvcontains the list of currently accepted taxonomic names for sea cucumbers.
This is real raw data with errors and inconsistencies. The first dataset is quite messy as it gathers information across several institutions, and for some of them, the specimens haven’t been examined in many years and may be identified with a taxonomic name that is not currently valid.
Before we get started, let’s get organized. This time, you will each work within your own GitHub repository. I already created them for you but they are empty.
- Start RStudio, and create a new project (File > New Project)
- Choose “Version Control”, and then “Git”
- In repository URL type:
https://github.com/r-bio/challenges-yourfirstname(e.g., mine ishttps://github.com/r-bio/challenges-francois) and choose a convenient location on your hardrive. - Create a new folder inside your working directory called
data. You can do this using the “New Folder” icon in the File panel in RStudio or by typingdir.create("data")at the R console. - Create a new script file (File > New File > R script) and save it as
cuke-challenge-firstname.R
Now, download the two data files inside your newly created data folder by
typing in your script file:
download.file("http://r-bio.github.io/data/holothuriidae-specimens.csv",
"data/holothuriidae-specimens.csv")
download.file("http://r-bio.github.io/data/holothuriidae-nomina-valid.csv",
"data/holothuriidae-nomina-valid.csv")
Save your script, and click on the Git icon below the menu, and choose “Commit”. Check the boxes in the “Staged” column for the all the files and write a commit message such as “add data”. Click on the “Commit” button, and then the “Push” icon. At least for me, at this stage I get an error message saying:
error: unable to read askpass response from 'rpostback-askpass'
This is apparently a known bug of RStudio (that I hope will be fixed soon). If you get it, close the “Review Changes” window, and in RStudio, in the menu go to Tools > Shell. There type:
git push -u origin master
it might ask for your GitHub username and password. Once it’s done, we won’t have to do this again and will be able to use the RStudio interface directly. If you have any problem at this stage, open an issue in the “logistics” repository and I’ll try to help you.
Now use the function read.csv() to load these datasets in memory. We’ll call
hol the data frame that contains the information about the specimens, and
nom the data frame that contains the information about the validity of the
species names.
hol <- read.csv(file="data/holothuriidae-specimens.csv", stringsAsFactors=FALSE)
nom <- read.csv(file="data/holothuriidae-nomina-valid.csv", stringsAsFactors=FALSE)
- How many specimens are included in the data frame
hol? - The column
dwc.institutionCodein theholdata frame lists the museum where the specimens are housed:- How many institutions house specimens?
- Draw a bar plot that shows the contribution of each institution
- The column
dwc.yearindicates when the specimen was collected:- When was the oldest specimen included in this data frame collected ? (hint: It was not in year 1)
- What proportion of the specimens in this data frame were collected between the years 2006 and 2014 (included)?
- The function
nzchar()on a vector returnsTRUEfor the positions of the vectors that are not empty, andFALSEotherwise. For instance,nzchar(c("a", "b", "", "", "e"))would return the vectorc(TRUE, TRUE, FALSE, FALSE, TRUE). The columndwc.classis supposed to contain the Class information for the specimens (here they should all be “Holothuroidea”). However, it is missing for some. Use the functionnzcharto answer:- How many specimens do not have the information for class listed?
- For the specimens where the information is missing, replace it with the information for their class (again, they should all be “Holothuroidea”).
- Using the
nomdata frame, and the columnsSubgenus.currentandGenus.current, which of the genera listed has/have subgenera? - We want to combine the information included in the
nomand theholspreadsheets, to identify the specimens in the data frame that use species names that are not valid. We’ll do this using the functionmerge(). By defaultmerge()only returns the rows for which there is an exact match in both datasets. Here, becausenomonly includes the names of the valid species, the results would not include any of the specimen information that do not have valid names. Read the help of themerge()function to learn more about it.- With the function
paste(), create a new column (calledgenus_species) that contains the genus (columndwc.genus) and species names (columndwc.specificEpithet) for theholdata frame. - Do the same thing with the
nomdata frame (using the columnsGenus.currentandspecies.current). - Use
merge()to combineholandnom(hint: you will need to use theall.xargument, read the help to figure it out, and check that the resulting data frame has the same number of rows ashol). - Create a data frame that contains the information for the specimens
identified with an invalid species name (content of the column
StatusisNA)? (hint: specimens identified only with a genus name shouldn’t be included in this count.) - Select only the columns:
idigbio.uuid,dwc.genus,dwc.specificEpithet,dwc.institutionCode,dwc.catalogNumberfrom this data frame and export the data as a CSV file (using the functionwrite.csv) namedholothuriidae-invalid.csv
- With the function
Once you are done, commit your script to your repository: Git icon > “Commit”, check the box next to the file name for your script, add a message, click on the Commit button and then on the “push” button.
Answers
## How many specimens?
nrow(hol)
## [1] 2984
## How many institutions house specimens?
length(unique(hol$dwc.institutionCode))
## [1] 4
## Barplot that shows the contribution of each institution:
barplot(table(hol$dwc.institutionCode))
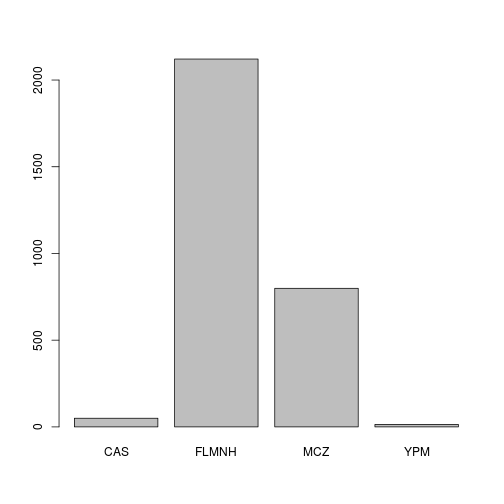
## When was the oldest specimen collected?
min(hol$dwc.year[hol$dwc.year > 1700], na.rm=TRUE)
## [1] 1902
## What is the proportion of speicmens collected between 2006 and 2014
sum(hol$dwc.year >= 2006 & hol$dwc.year <= 2014, na.rm=TRUE)/nrow(hol) # for all specimens
## [1] 0.4932976
sum(hol$dwc.year >= 2006 & hol$dwc.year <= 2014, na.rm=TRUE)/sum(!is.na(hol$dwc.year)) # for all specimens with a year
## [1] 0.6986236
## How many specimens are missing the "Class" data?
sum(!nzchar(hol$dwc.class))
## [1] 50
## Add the missing data
hol$dwc.class[!nzchar(hol$dwc.class)] <- "Holothuroidea"
## Which of the genera listed have subgenera?
unique(nom$Genus.current[nzchar(nom$Subgenus.current)])
## [1] "Holothuria"
## Combine the two data frames
hol[["genus_species"]] <- paste(hol$dwc.genus, hol$dwc.specificEpithet)
nom[["genus_species"]] <- paste(nom$Genus.current, nom$species.current)
hol_combined <- merge(hol, nom, all.x=TRUE)
nrow(hol_combined) == nrow(hol)
## [1] TRUE
## How many specimens are identified with currently invalid species names?
hol_invalid <- subset(hol_combined, is.na(Status) & nzchar(dwc.specificEpithet))
write.csv(hol_invalid[, c("idigbio.uuid", "dwc.genus", "dwc.specificEpithet", "dwc.institutionCode", "dwc.catalogNumber")],
file="holothuriidae-invalid.csv", row.names=FALSE)